


















 539 / 658
539 / 658

3
High-Resolution LC-MS/MS
High-resolution LC-MS/MS analysis was carried out using a
Thermo Scientific
™
EASY-nLC
™
system and Thermo
Scientific
™
LTQ Orbitrap XL
™
hybrid ion trap-Orbitrap
mass spectrometer. Samples in 5% (vol/vol)
acetonitrile/0.1% (vol/vol) formic acid were injected into a
Thermo Scientific
™
Hypersil GOLD
™
aQ fused-silica
capillary column (75 µm x 25 cm, 5 µm particle size) in a
250 µL/min gradient of 5% acetonitrile/0.1% formic acid to
30% acetonitrile/0.1% formic acid over the course of
180 minutes. The total run time was 240 minutes and the
flow rate was 285 nL/min. The LTQ Orbitrap XL MS was
operated at 60,000 resolution (FWHM at
m/z
400) for a
full scan for data-dependent Top 5 MS/MS experiments
(CID or HCD). The top 5 signals were selected with
monoisotopic precursor selection enabled, and +1 and
unassigned charge states rejected. Analyses were carried out
in the ion trap or the Orbitrap analyzer. The experiments
were performed using collision-induced dissociation (CID)
and higher-energy collisional dissociation (HCD)
fragmentation modes.
SRM Methods
SRM methods were developed on a Thermo Scientific
™
TSQ Vantage
™
triple stage quadrupole mass spectrometer
with a Thermo Scientific
™
Accela
™
pump, a CTC PAL
®
autosampler (Leap Technologies), and a Thermo Scientific
™
Ion Max
™
source equipped with a high-flow metal needle. A
mass window of 0.7 full width at half maximum (FWHM,
unit resolution) was used in the SRM assays because the
immunoenriched samples had a very high signal-to-noise
ratios. Narrower windows were necessary when the matrix
background was significant and caused interferences that
reduced signal-to-noise in the SRM channels. Reversed-
phase separations were carried out on a Hypersil GOLD
column (1 mm x 100 mm, 1.9 µm particle size) with a flow
rate of 160 µL/min. Solvent A was 0.2% formic acid in
LC-MS-grade water, and solvent B was 0.2% formic acid in
Fisher Scientific
™
Optima
™
-grade acetonitrile.
Software
Thermo Scientific
™
Pinpoint
™
software was used for
targeted protein quantification, automating the prediction of
candidate peptides and the choice of multiple fragment ions
for SRM assay design. Pinpoint software was also used for
peptide identity confirmation and quantitative data
processing. The intact PTH sequence was imported into the
software and digested with trypsin
in silico
. Then, transitions
for each peptide were predicted and tested with recombinant
PTH digest to determine those peptides and transitions
delivering optimal signal. After several iterations, a subset of
six peptides with multiple transitions was chosen.
Further tests were conducted with this optimized method.
After the target peptides were identified, heavy arginine or
lysine versions were synthesized to be used as internal
quantitative standards. Target peptides were subsequently
identified and quantified by coeluting light- and heavy-
labeled transitions in the chromatographic separation. Time
alignment and relative quantification of the transitions were
performed with Pinpoint software. All samples were assayed
in triplicate.
Results and Discussion
Top-Down Analysis and Discovery of Novel Variants
The approach described herein coupled targeting a common
region of PTH by use of a polyclonal antibody (raised to the
C-terminal end of the protein) with subsequent detection by
use of SRMMS. Numerous PTH variants were
simultaneously extracted with a single, high-affinity
polyclonal antibody, and the selection of the epitope was
directed by the target of interest (i.e., intact and N-terminal
variants). The primary goal was to differentiate between
intact PTH1–84 and N-terminal variant PTH7–84 while
simultaneously identifying any additional N-terminal
heterogeneity throughout the molecule. The results of these
top-down experiments allowed the development of an initial
standard profile for PTH. Clearly, this profile is not finite,
and may be expanded to include additional variants found
through literature search and/or complementary full-length
studies. However, this standard profile provided an initial
determination of target sequences for developing specific
SRM assays.
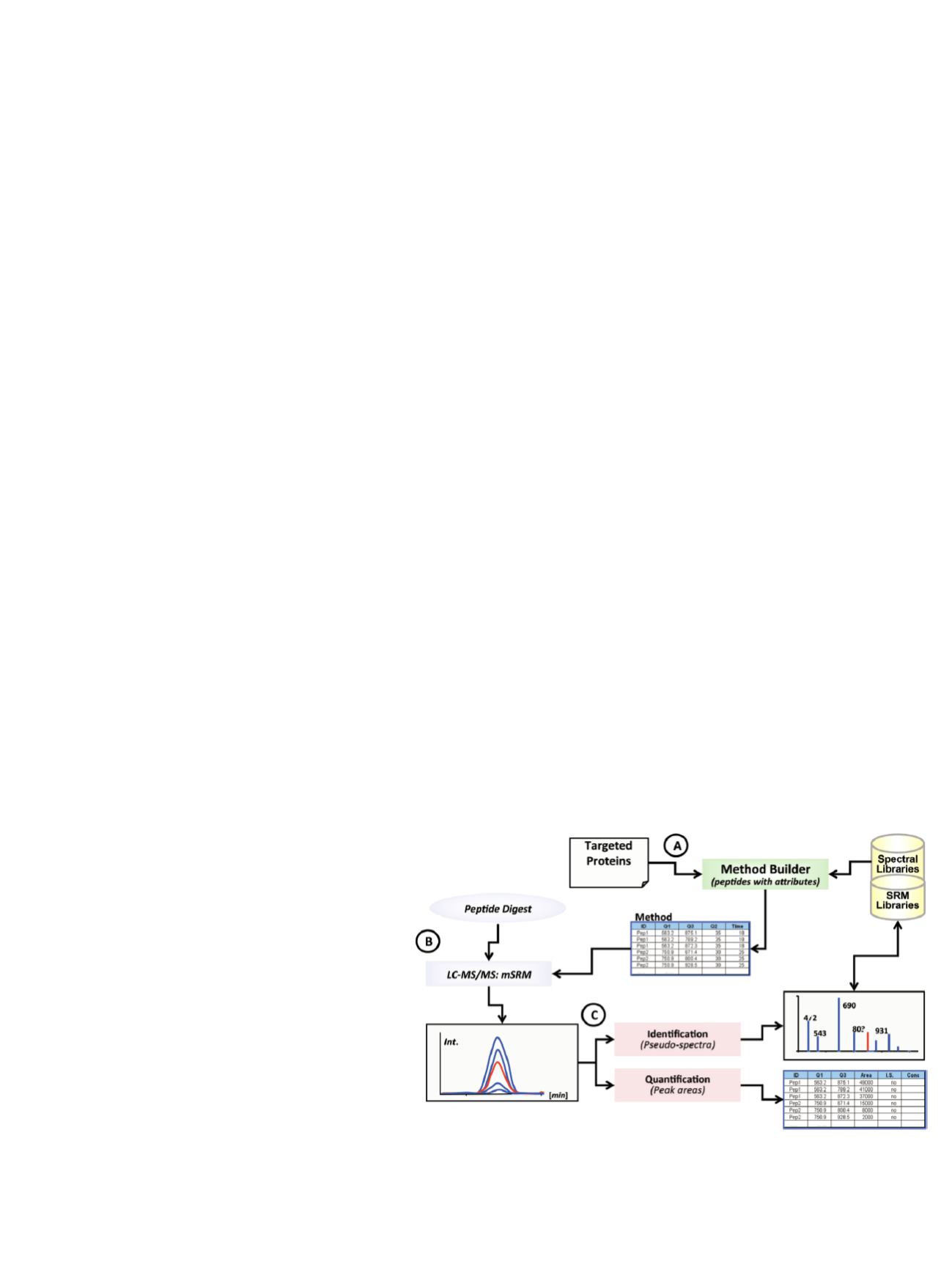
Selection of Transitions for SRM
During LC-MS/MS analysis, multiple charge states and
fragmentation ions were generated from each fragment,
resulting in upwards of 1000 different precursor/product
transitions possible for PTH digested with trypsin. Empirical
investigation of each transition was not efficient. Therefore,
a workflow incorporating predictive algorithms with
iterative optimization was used to predict the optimal
transitions for routine monitoring of tryptic fragments
(Figure 2). The strategy facilitated the translation of peptide
intensity and fragmentation behavior empirically obtained
by high-resolution LC-MS/MS analyses to triple quadrupole
SRM assays. Inherent to the success of the workflow was
the similarity of peptide ion fragmentation behavior in these
ion trap and triple quadrupole instruments.
12
Empirical data
from such LC-MS/MS experiments were used in conjunction
with computational methods (
in silico
tryptic digestions and
prediction of SRM transitions) to enhance the design of
effective SRM methods for selected PTH peptides.
Figure 2. Pinpoint workflow for development of multiplexed SRM assays.
[Q = quadrupole; mSRM = multiple SRM; Int. = intensity; I.S. = internal standard;
Conc = concentration. Time measurements are in minutes (min).]