


















 151 / 302
151 / 302

3
Moving to the Next Step Beyond Triple Quadrupole MS/MS Quantitation
The Thermo Scientific Application Note 63552
5
describes a quick and simple method for biotoxin
analysis based on the EURL LC-MS protocol.
6
Utilizing offline sample preparation and
determination by the Thermo Scientific
™
TSQ Quantum Ultra
™
triple quadrupole mass
spectrometer, the method meets general performance criteria required for the analysis of lipophilic
shellfish toxins. The simplicity of the method makes this approach suitable for any routine lab
doing the analysis.
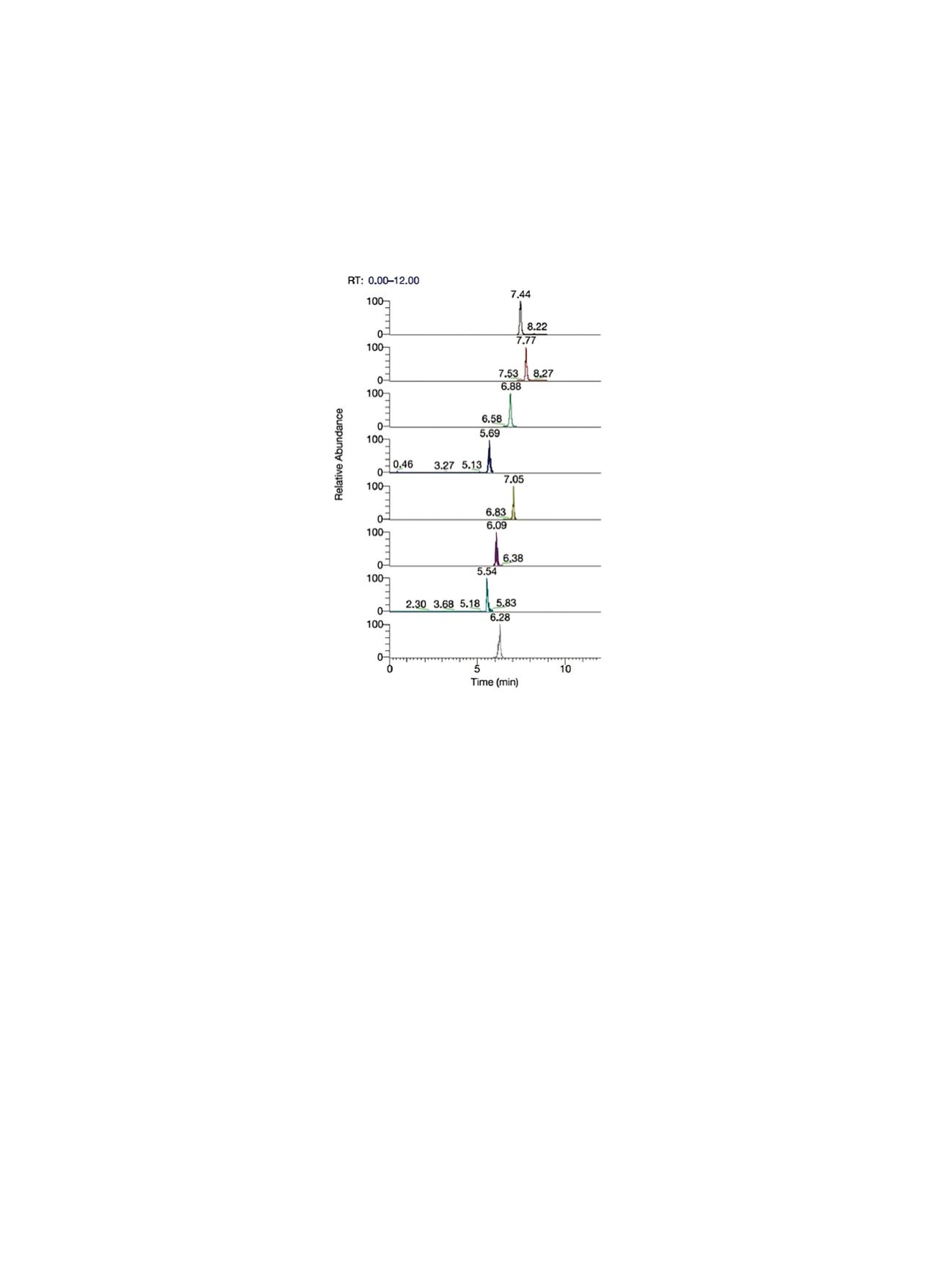
Figure 1 documents the selectivity of the method by showing the extracted ion chromatograms
for the analyzed compounds at 40 μg/kg levels. As can be seen, the selectivity of the MS/MS mass
spectrometer allows easy detection of all compounds at levels required by regulation.
Figure 1. Chromatogram of sample containing 40 ppb of toxins, analyzed by TSQ Quantum Ultra quadrupole LC-MS
(Retention Time: 7.44 min – AZA-1; 7.77 min – AZA-2; 6.88 min – AZA-3; 5.69 min – OA; 7.O5 min – DTX-1; 6.06 min – DTX-2;
5.54 min – YTX; 6.28 min – PTX-2).
In general, the utilization of triple quadrupole LC-MS/MS is a viable solution for any lab
performing targeted analysis of the well-defined groups of compounds. It is, however, not
suitable for the analysis of a broader range of compounds, metabolites or conjugates for which
the structural information is not known.
Future Approaches: Accurate Mass Data
As already mentioned, in response to the need for non-targeted methods that can potentially detect
unknowns, metabolites or adducts, HRAM has been successfully implemented for screening and
quantification in food safety applications. The lower cost, higher mass accuracy, and ease-of-use
of modern quadrupole time-of-flight (QTOF) and Orbitrap—based mass spectrometers have
made high-resolution systems viable alternatives to triple quadrupole systems for routine analysis.
After full-spectrum data acquisition, specificity is typically achieved by extracting narrow mass
windows (ie. 2–5 ppm) centered around a list of target analytes. Using this approach, it has been
demonstrated that a resolving power of 50,000 or greater is required for correct mass assignments
in complex matrices.
3